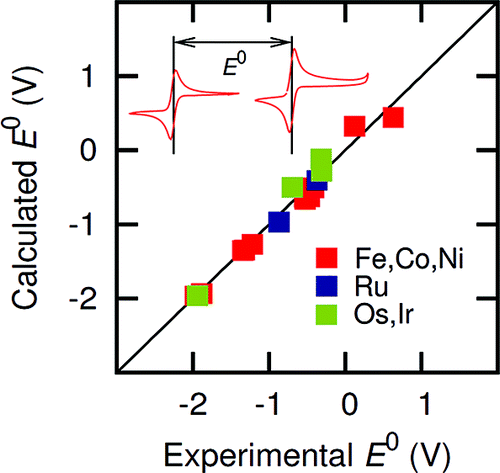
Reliable calculations of redox potentials could provide valuable insight into catalytic mechanisms of electrochemically active transition-metal complexes as well as guidelines for the design of new electrocatalysts. However, the correlation between theoretical and experimental data is often uncertain, since redox properties depend strongly on experimental conditions of electrochemical measurements, including the nature of the solvent, electrolyte, and working electrode. Here, we show that the use of internal references allows for quantitative theoretical predictions of redox potentials with standard deviations σ comparable to typical experimental errors of cyclic voltammetry measurements. Agreement for first-, second-, and third-row transition-metal complexes is demonstrated even at a rather modest level of density functional theory (σ = 64 mV for the UB3LYP/6-311G* level). This is shown for a series of benchmark redox couples, including ([MCp2]0/+ (Cp = η5-cyclopentadienyl), [MCp*2]0/+ (Cp* = η5-1,2,3,4,5-pentamethylcyclopentadienyl), [M(bpy)3]2+/3+ (bpy =2,2′-bipyridine), and [Ir(acac)3]0/+ (acac = acetylacetonate), with M = Fe, Co, Ni, Ru, Os, or Ir) in various nonaqueous solvents [acetonitrile (MeCN), dimethyl sulfoxide (DMSO), and dichloromethane (DCM)].